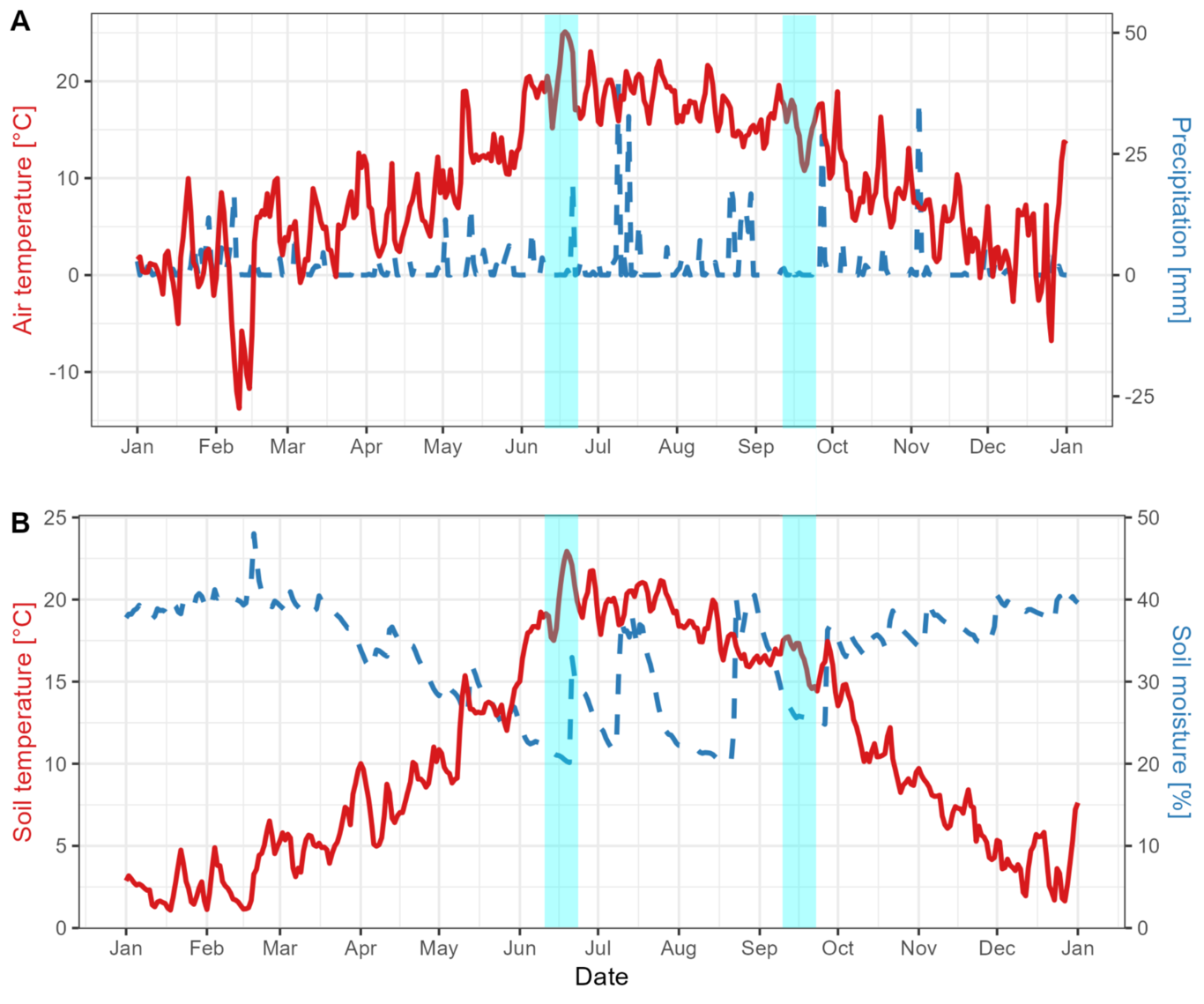
INTRODUCTIONCompelling evidence shows that biodiversity enhances essential ecosystem functions, such as productivity and decomposition rates (Loreau & Hector 2001; Hooper et al., 2005; Cardinale et al., 2012). One primary underlying reason may be that individual species or groups of species in different functional groups may have dissimilar niches (niche complementarity effects ) which allow diverse communities to maximize resource utilization and minimize competition (Cardinale et al., 2011; Zuppinger-Dingley et al., 2014). In theory, such niche differences include temporal variation in biological activity (Ebeling et al., 2014), and species in a community can adjust the timing of their biological activity in such a way that they cover the longest possible time and/or use the resources from the largest possible space in the habitat. If phenological niche differences are high enough, they can affect the shape of the phenology at the community level. For instance, if a plant community is composed of species that grow in early spring, the aboveground growing season will be extended, compared with a community lacking those species (Ebeling et al., 2014; Rudolf, 2019). Therefore, species and functional group diversity can affect the timing of community-level productivity (i.e. community phenology) via temporal niche differentiation and/or increasing the probability of species with those traits to occur in the community (selection effect ) (Loreau & Hector 2001). However, variation in phenology is primarily monitored at the species rather than community level. Moreover, phenological variation is typically attributed to changes in climate drivers, such as temperature and water supply (Wright and van Schaik 1994; Staggemeier et al., 2018), and has rarely been quantified as a response to changes in biodiversity (but see Wolf et al., 2017 and Guimarães-Steinike et al., 2019).Most ecosystem processes are soil-related or even soil-dependent (Bardgett & van der Putten, 2014; Soliveres et al., 2016; Schuldt et al., 2018). However, phenology tends to be monitored on easily observed aboveground response variables, and evidence describing soil phenology is mostly lacking (Bonato Asato et al., 2023). This knowledge gap leads to uncertainty about how well soil properties and belowground processes (i.e. root growth and activity of soil organisms) are predicted by aboveground phenological strategies (Eisenhauer, 2012; Blume-Werry et al., 2015; Eisenhauer et al., 2018). Because shoots and roots are interdependent, tight synchrony of their responses to environmental drivers is often expected (Iversen et al., 2015; but see Blume-Werry et al., 2016). However, the role of biotic and abiotic constraints on this synchrony seems to vary significantly among ecosystems and plant types, ultimately affecting which organs grow first, faster, or remain active and alive longer. Moreover, plant (roots and shoots) processes are often assumed to indicate ecosystem functions driven by the activity of organisms at adjacent trophic levels, such as soil fauna, but this may not necessarily be the case. Hot moments (within-year events inducing high activity) in soil organism activity depend, in part, on inputs from root exudates or pulses of detrital inputs from senescent roots (Kuzyakov & Blagodatskaya 2015). However, the limited evidence from the field does not always confirm plant-activity-based assumptions. For example, phenological monitoring of detritivore feeding activity during the growing season in oaks has shown both a negative and no correlation between feeding activity and oak branch production (Eisenhauer et al., 2018). In an experimental grassland, feeding activity rates decreased during the summer, when plant growth is usually high (Siebert et al., 2019; Sünnemann et al., 2021). Evidence suggests that investments in shoot and root production are commonly not synchronous (e.g. Steinaker & Wilson 2008; Steinaker et al. 2010; Sloan et al. 2016; Blume-Werry et al. 2016), as well as the dynamics of soil organisms (Bonato Asato et al., 2023; Eisenhauer et al., 2018). However, we lack experimental evidence demonstrating whether changes in biodiversity may influence the predictability and synchronization of the dynamics above and below the ground.Presently, two predominant conceptual frameworks delineate the interplay between biodiversity and the synchronization of ecosystem functions. On the one hand, ecosystem stability theory suggests that increasing biodiversity increases temporal asynchrony among populations and functions, which would be one of the primary mechanisms for positive diversity-stability relationships (Cardinale et al., 2013; Loreau & de Mazancourt 2013). In other words, temporal asynchrony is needed for a healthy (stable) ecosystem functioning. On the other hand, ecosystem coupling, as defined by Ochoa-Hueso et al. (2021) as ”the orderly connections between the biotic and abiotic components of ecosystems across spaces and/or time”, suggests the opposite: for more efficiently process, cycle, and transfer of energy and matter, a higher temporal coupling of populations and functions is needed. Under this point of view, temporal synchrony is required for more efficient ecosystem functioning, and monitoring the dynamics of one function or population can be used as an indicator of activity in the other. In both cases, disruptions such as biodiversity change, may affect key aboveground or belowground processes, leading to acceleration or delay of community phenology and desynchronization of ecosystem functions. Despite the potential importance of aboveground-belowground phenological synchrony, the current lack of studies concurrently monitoring shoot, root, and soil fauna dynamics has impeded a thorough understanding of the mechanisms by which changes in biological diversity may influence the responses of these affiliated processes.Here, we examine how experimentally manipulated plant diversity influences the phenological patterns of shoot, root, and soil fauna dynamics (responses). In the framework of a long-term grassland biodiversity experiment (the Jena Experiment; Roscher et al. 2014; Weisser et al. 2017), using well-established methods (LiDAR, phenological cameras, minirhizotrons, bait-lamina strips), we measure ecosystem response variables that are often used to evaluate aboveground-belowground ecosystem functioning and biological activity in annual plant communities (e.g. plant community height, greenness, root production, and detritivore feeding activity) every two to three weeks over four seasons (one full year). We used these data to calculate yearly values for each response variable, phenological patterns, and synchrony between response variables. With this approach, we ask the following questions:1) How does plant diversity affect the yearly accumulated values of aboveground plant traits and belowground activity? We expect that increasing plant diversity throughout the year enhances all response variables (Weisser et al. 2017; Mommer et al., 2015; Eisenhauer et al., 2010).2) Does plant diversity affect intra-annual aboveground and belowground phenological patterns? We predict that plant community shoot dynamics will be concentrated in spring and summer, as usual in temperate regions. Root production should last longer than that of shoots, as found in other studies (Steinaker & Wilson 2008; Blume-Werry et al. 2016), even though it is not clear if this longer activity is driven by an earlier start of the production, a later end, or both. For detritivore feeding activity, we expect a peak in early spring due to high moisture and increased temperature and another peak in autumn, driven by the increased availability of resources by above- and belowground plant-derived inputs and high moisture.3) Do changes in plant diversity affect the synchrony of shoot, root, or soil organism dynamics? We expect plant species richness and functional group richness to enhance aboveground-belowground activity, which could lead to either more or less synchronized patterns. If plant diversity drives enhanced functioning at different time points (e.g. advances plant growth and delays root senescence), we could see a negative diversity effect on synchrony.4) Does the time of year influence the strength/direction/predictability of relationships between aboveground-belowground response variables? Because plant shoots are only active for a restricted period, we expect plant diversity effects to be most pronounced during the growing season (Guimarães-Steinike et al., 2019), while abiotic constraints might mostly drive belowground dynamics out of the growing season.


/screenshot (1).png?1566398100)